Amplicon analysis
amplicon analysis
WARNING!
Don't get too lost in the weeds when working with marker-gene data. More often than not the kind of detailed questions that will land you in that situation can't be answered with this type of data anyway!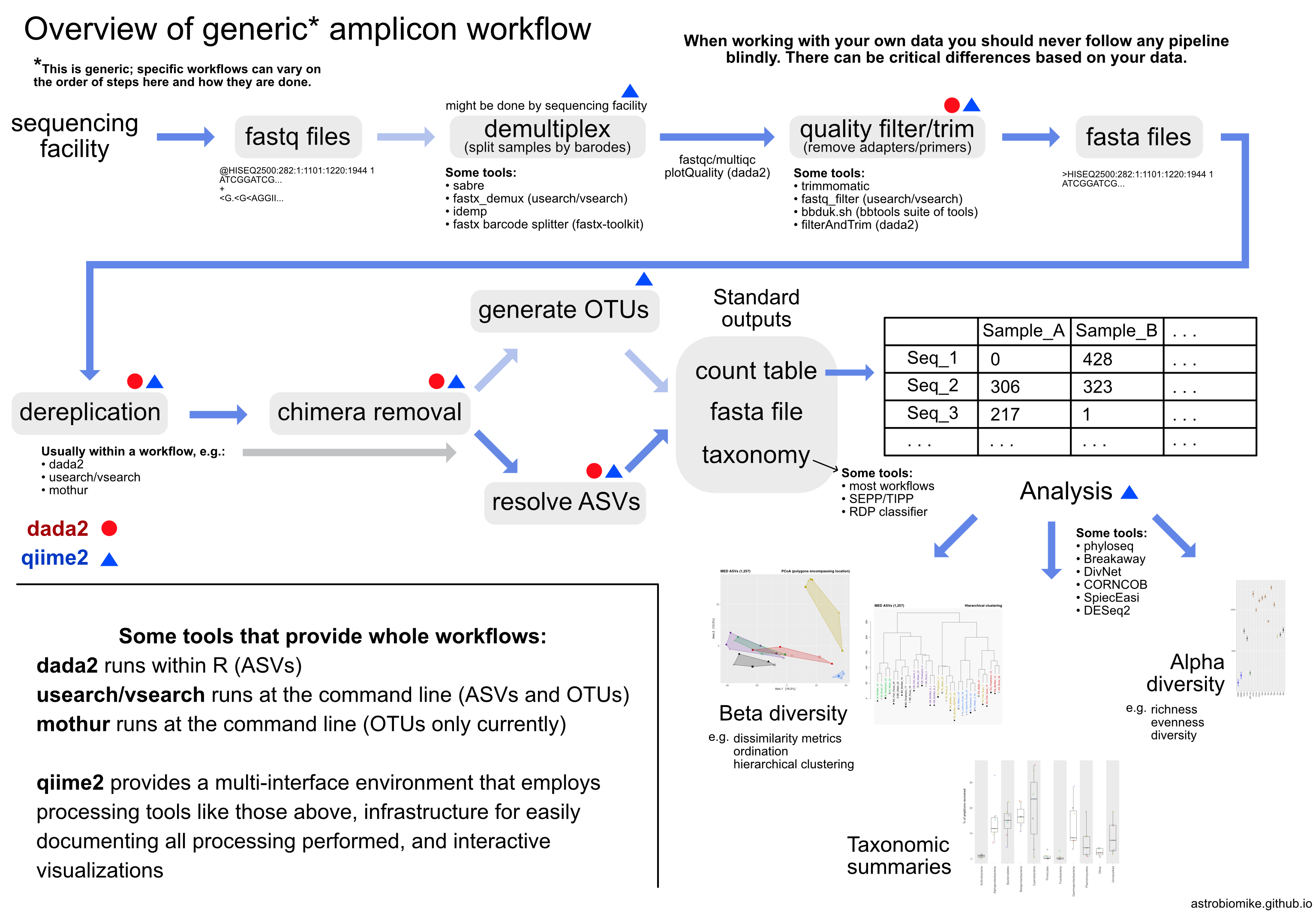
Amplicon pages
- Demultiplexing example
- A full walkthrough of processing and initial analyses
- An example of dealing with 16S and 18S mixed together
See this page for an overview of some of the differences between amplicon sequencing and metagenomics.
Amplicons and marker-genes and tags, oh my!
Most often a marker-gene analysis is the microbial ecologist’s first tool in a vast toolkit. It is primarily used as a broad survey of community structure. As the warning notes above, when first beginning to work with this type of data it can be easy to get caught spinning your wheels about a subtle component in your processing pipeline that ultimately has a negligible impact compared to the noise we are working through. What I mean by this is, generally speaking, tag sequencing is most often not the appropriate tool to answer really meticulous questions. It is a tool for comparing baseline proxies of metrics about microbial communities. It is a tool of exploration and hypothesis generation.
ASVs vs OTUs There are many ways to process amplicon data. Some of the most widely used tools/pipelines include mothur, usearch, vsearch, Minimum Entropy Decomposition, DADA2, and qiime2 (which employs other tools within it). If you are looking solely at a broad level, you will likely get very similar results regardless of which tool you use so long as you make similar decisions when processing your sequences (e.g. decisions about things like minimum abundance filtering), so don’t get too lost in the weeds of trying to find the “best” tool for processing amplicon data. That said, there is a movement in the community away from the traditional OTU approach and on to single-nucleotide-resolving methods that generate what are referred to as ASVs (amplicon sequence variants). And the reasoning for this is pretty sound, as recently laid out very nicely by Callahan et al. here, but the intricacies of the differences may seem a little nebulous at first if you’re not used to thinking about these things yet. If you are new to this, know that most of the experts on these things would absolutely recommend using a newer method that resolves ASVs (enables single-nucleotide resolution) over a more traditional OTU approach (like 97% or 99% OTU clustering). It may be the case that you’d like a broader level of resolution than what initial ASVs will give, but it is still best to first generate ASVs and then you can always “back out” your resolution with clustering at some threshold or binning sequences by taxonomy or whatever. This is because the underlying unit, the inferred ASV, is most often more biologically meaningful and a more useful unit beyond the current dataset than the units produced by traditional OTU clustering methods. With all of that said, if I were processing a new amplicon dataset, I would currently use DADA2, and there is a full example workflow using that here.
Some terminology
As with most things, trying to pin down precise, one-answer definitions can be difficult sometimes. This is because language is fluid and we are constantly trying to find better ways to communicate more efficiently and with more clarity. That’s a good thing, but it can add to the confusion when first trying to find your footing in a new area. Here are some terms you might hear and what they mean most often in my experience (though there can certainly be other interpretations).
Amplicon
The resulting sequence of a targeted amplification of genetic material. Targeted meaning primers were used
Marker gene
A gene that can be useful for delineating organisms, like a fingerprint.
OTU
Operational Taxonomic Unit. OTUs are an artificial, arbitrary construct useful for grouping sequences together into units to help us summarize and analyze things, and they also intended to help deal with sequencing error intrinsic to the technology.
ASV
Amplicon Sequence Variant. Resulting sequences from newer processing methodologies that attempt to take into account sequencing error rates and are believed to represent true biological sequences. The case for moving towards using ASVs over OTUs is made very well in this paper by @bejcal, @joey711, and @SherlockpHolmes.
Tag sequencing
In my opinion, the most useful and most often used meaning of “tag sequencing” is when you are using primers that target something specific, like a marker gene. So the opposite of shotgun sequencing which uses random hexamer or nonamer primers. But on rare occasions I have heard this used to mean barcoding samples.
Barcodes
Barcodes refer to the sequences ligated to your individual samples’ genetic material before they get all mixed together to be sequenced together. These barcodes are then unique to each sample, so you can afterwards identify which sequences came from which samples.
Demultiplex
Demultiplexing refers to the step in processing amplicon sequence data where you’d use the barcode information in order to know which sequences came from which samples after they had all been sequenced together.