Demultiplexing
Demultiplexing
amplicon analysis
The DADA2 and u-/v-search tutorials on this stie both take off assuming your samples have already been demultiplexed. Demultiplexing refers to the step in processing where you’d use the barcode information in order to know which sequences came from which samples after they had all be sequenced together. Barcodes refer to the unique sequences that were ligated to your each of your invidivual samples’ genetic material before the samples got all mixed together. Depending on your sequencing facility, you may get your samples already split into individual fastq files, or they may be lumped together all in one fastq file with barcodes still attached for you to do the splitting. If this is the case, you should also have a “mapping” file telling you which barcodes correspond with which samples. Here we’re going to cover one way in which to demultiplex your samples.
Tools used here
There are many freely available tools to perform demultiplexing. Here I’ll demonstrate with Sabre. Sabre is awesomely simple and quick, and it is installable through the wonderful environment manager Conda 🙂
Once we have conda, an environment with sabre can be created like so:
conda create -n sabre -c conda-forge -c bioconda -c defaults sabre
# and activating it
conda activate sabre
The data
You certainly don’t need to download this example data, but just in case you wanted to, you can from here (it’s tiny, 30 Mb):
cd ~
curl -L -o demultiplex_ex.tar.gz https://ndownloader.figshare.com/files/11461430
tar -xzvf demultiplex_ex.tar.gz
rm demultiplex_ex.tar.gz
cd demultiplex_ex
Formatting the mapping file
In this example’s case the sequencing was performed by MR DNA. Their mapping file (at the time these samples were sequenced) looks like this:
column -t 100914ML515F-mapping.txt | head
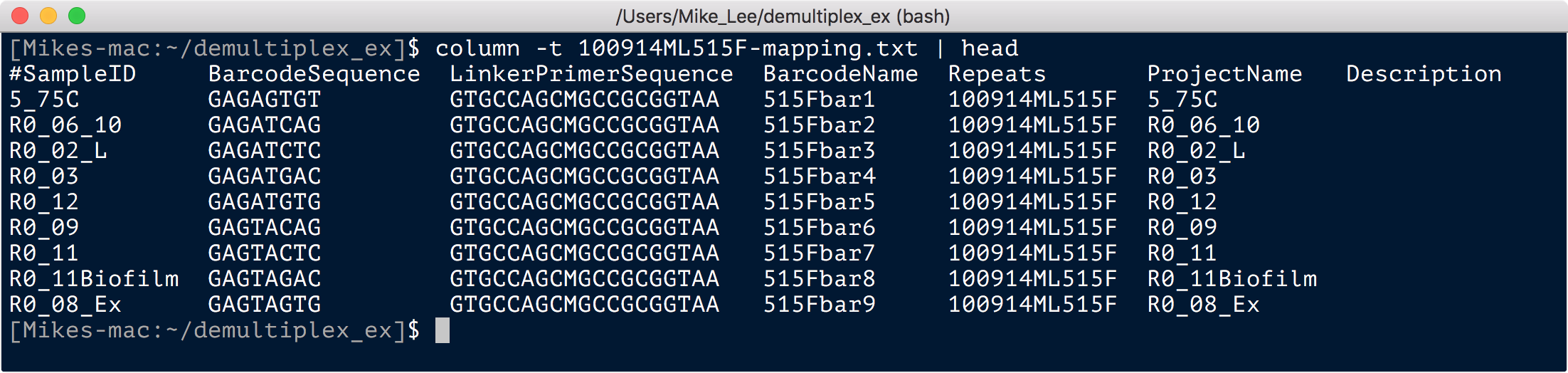
Here all we really care about for demultiplexing are this first and second columns, the sample name and the barcode. The sabre formatting required, as laid out here on their github, wants 3 tab-delimited columns: 1) barcode; 2) name for forward read file; 3) name for reverse read file. So one easy way to make this file is with the magic of Unix:
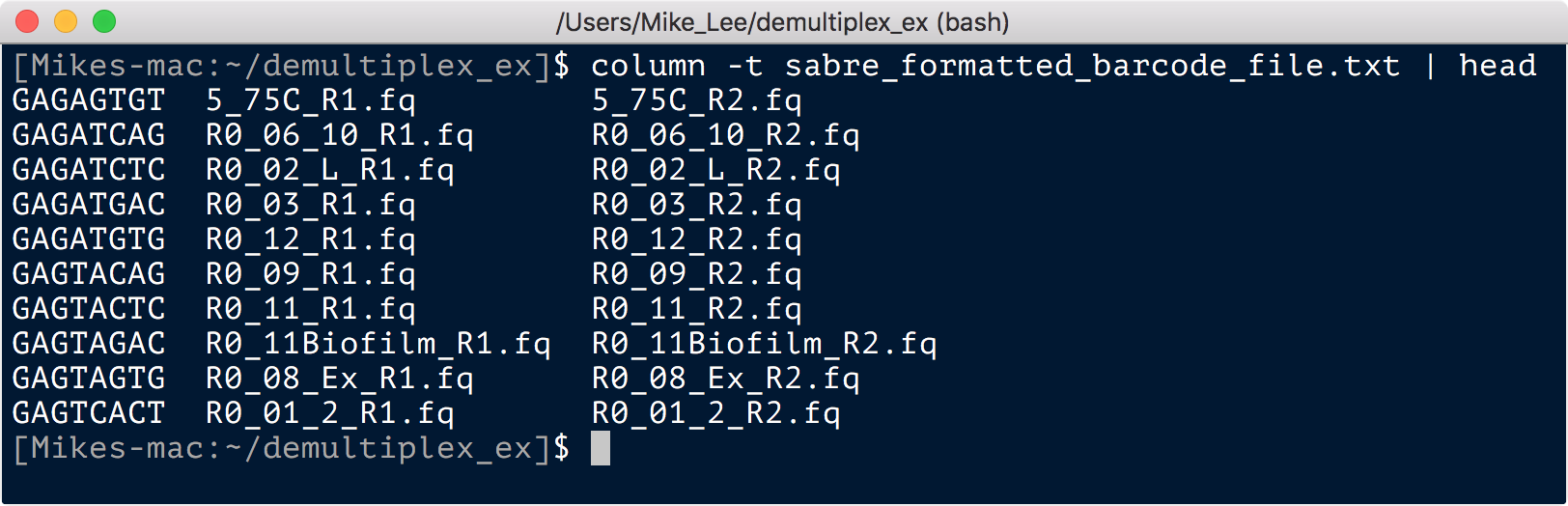
awk -v OFS="\t" ' NR > 1 {print $2, $1"_R1.fq", $1"_R2.fq"} ' 100914ML515F-mapping.txt > sabre_formatted_barcode_file.txt
Here we are using awk to skip the first row of headers with NR > 1, and then print the columns as we specify them (appending to the sample names “_R1.fq” and “_R2.fq”, and in the order we specify them, to a new file that is tab-delimited – set with the OFS="\t":
If that awk command doesn’t make any sense to you and being able to use Unix to manipulate files like this would be useful to your work, then I highly recommend running through the Unix crash course to get a better grasp on things 🙂
Demultiplexing with Sabre
Now that we have the mapping file formatted appropriately for what Sabre wants, running it is cake. In this case we have paired end fastq files, but there are other usage examples here. Here the -f flag is for the forward read, -r for reverse, -b for our mapping file, -u for forward reads that didn’t match a barcode (Sabre by default allows no mismatches), and -w for reverse reads that didn’t match:
sabre pe -f Sam78-125_S3_L001_R1_001.fastq -r Sam78-125_S3_L001_R2_001.fastq -b sabre_formatted_barcode_file.txt -u no_bc_match_R1.fq -w no_bc_match_R2.fq
Sabre will spit out some info for how many reads were assigned to each barcode, and now there will be fastq files for each sample in your directory:
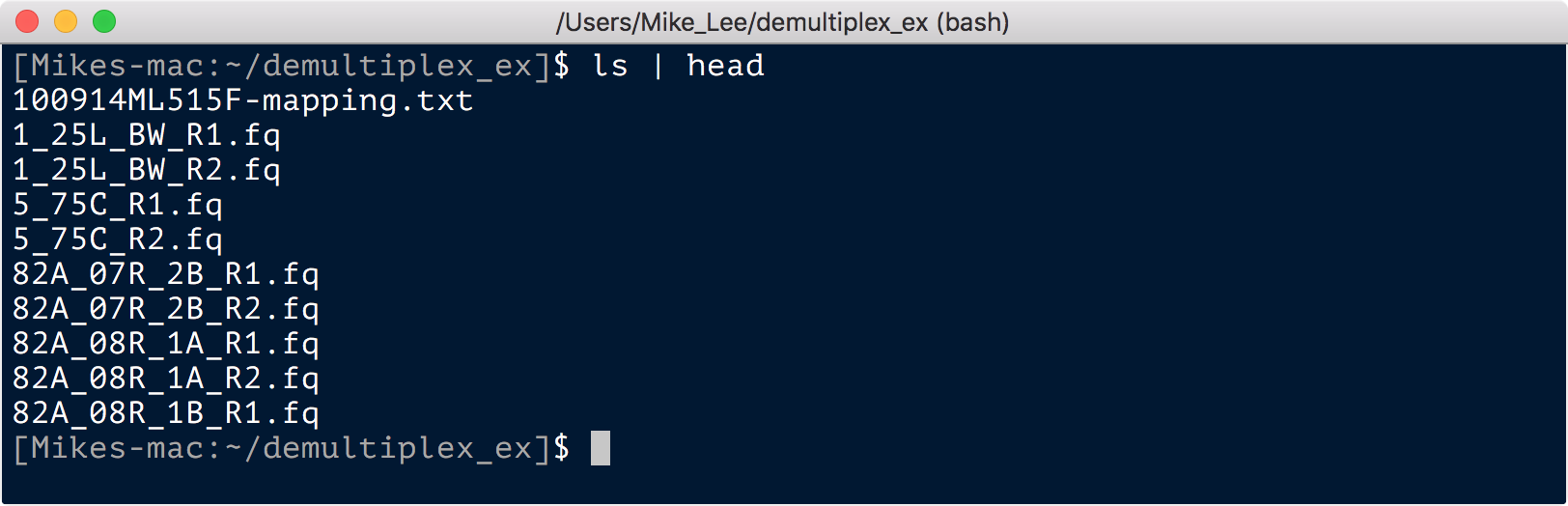
Sabre also removed the barcodes from the forward reads for the read pairs it was able to split. If your primers and sequencing protocol resulted in full overlap, you may have the reverse complement of the barcodes at the end of the reverse reads. Sabre has a -c flag for this, but on the couple of datasets I tried that on, it doesn’t seem to be operating in the same orientation my reverse reads were in. That is however dealt with if you use something like bbduk – which I have an example of in the DADA2 example workflow here – to remove the primers (which lie inside of the barcode in both reads).