Installing tools at the command line
Installing Tools
Unix
CONDA ALERT!
This page was created before conda became the wonderful powerhouse it is today. Conda is a package and environment manager that is by far the easiest way to handle installing most of the tools we would want to use.
There is an introduction to conda page here that I highly recommend looking over and trying first for any installation goals.
This page will stay here for archival purposes and to hopefully help if we run into something that conda can’t help with 🙂
ATTENTION!
The installation examples here all download and install things into a directory called~/happy_bin. If you are new to this stuff and want to be able to follow the examples here more closely, be sure to familiarize yourself with what the PATH is here and to follow these instructions in order to create that directory and add it to your PATH.As we’ve seen in the Unix crash course and real-life examples sections, you can do some amazing things with standard Unix commands as far as manipulating plain-text files goes. But that’s just the start of things. If you’re working towards more bioinformatic-leaning applications, you’re certainly also going to need to download and/or install lots of tools that don’t come standard. What’s required on our end to get something working properly varies by the tool, and most that are highly used by people have excellent documentation on how to properly download and install them. This page will be an ongoing list of the installation process for any of the tools used on tutorials from this site, and they will hopefully serve as examples to help guide you through installing other things. Many programs have differences between which version we’d need and how we’d install them on our personal computer vs installing on a server. I plan to add a section soon covering those differences, but for now these are examples for installing on a Mac personal computer and would need to be adjusted for other systems.
What’s a binary?
You may have heard or read an expression before referring to whether there is a binary or executable available for download for a particular program. Programs stored in these formats can just be downloaded and then utilized usually with no other work required – they are already “built” when you download them. This is in contrast to programs that you need to download and then “compile from source” – you need to “build” the program first in order for it to work. As usual there is a give and take here, binaries are easier to grab and get running, but you have more freedom and often access to the newest developments when installing something from its source code. There is a good explanation on stack exchange about some of these differences. The examples here so far are all binaries (as I’ve been selecting programs like that on purpose for ease of use in tutorials), but I will try to get an example up soon of installing from source.
First we need a happy bin
Often a big part of getting things to work properly is having them in a location on the computer that you can access no matter where you are. As we covered here, a list of directories that are scanned for programs automatically by your computer is stored in the special variable called “PATH”. For the sake of simplicity, many tools we use in tutorials on this site are going to be put in a directory called ~/happy_bin. You can check to see if this is in your PATH already like this:
echo $PATH | tr ":" "\n" | grep "happy_bin"
If this returns something like this:
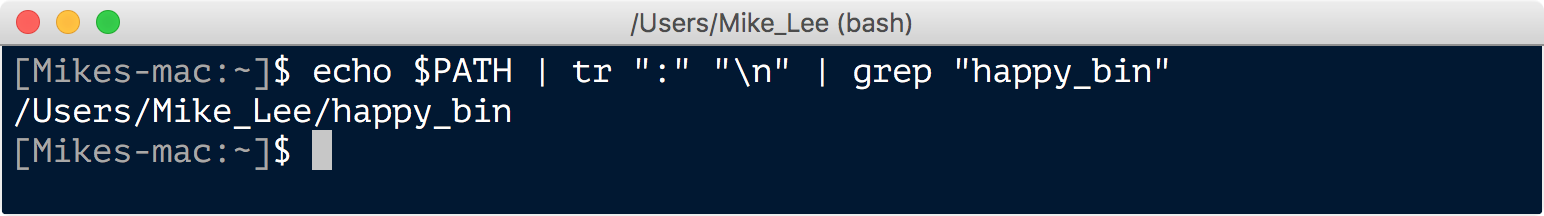
You’re good to go. If nothing was returned, then you can create this directory and add it to your PATH like such:
mkdir ~/happy_bin
echo 'export PATH="$PATH:~/happy_bin"' >> ~/.bash_profile
And if you’re unsure of what’s going on here, be sure to visit the modifying your PATH page.
Installing tools used on this site
As explained above, most things we install here will be put in a directory in our home location called ~/happy_bin, so pay attention to modify any code here accordingly if you want to put things somewhere else. If you’re unfamiliar with what’s going on in any of these code blocks below, and want to be more familiar, run through the Unix crash course and modifying your PATH pages sometime 🙂
My BioInformatics Tools (bit)
These are a collection of one-liners and short scripts I use frequently enough that it’s been worth it for me to have them instantly available anywhere. This includes things like:
- downloading NCBI assemblies in different formats by just providing accession numbers (
bit-dl-ncbi-assemblies) - pulling out sequences by their coordinates (
bit-extract-seqs-by-coords) - splitting a fasta file based on headers (
bit-parse-fasta-by-headers) - renaming sequences in a fasta (
bit-rename-fasta-headers) - pulling amino acid or nucleotide sequences out of a GenBank file (
bit-genbank-to-AA-seqs/bit-genbank-to-fasta)
And other just convenient things to have handy like removing those annoying soft line wraps that some fasta files have (bit-remove-wraps) and printing out the column names of a TSV with numbers (bit-colnames) to quickly see which columns need to be provided to things like cut or awk. Some require biopython and pybedtools, but all is taken care of if you use the the conda installation 🙂
Each command has a help menu accessible by either entering the command alone or by providing -h as the only argument. Once installed, you can see all available by entering bit- and pressing tab twice.
Conda install
conda install -c bioconda -c astrobiomike bit
Non-conda way
cd ~/happy_bin/
wget https://github.com/AstrobioMike/bioinf_tools/archive/master.zip
unzip master.zip
mv bioinf_tools-master bioinf_tools
echo 'export PATH="$PATH:~/happy_bin/bioinf_tools"' >> ~/.bash_profile
source ~/.bash_profile
Some of these require the python packages BioPython and pybedtools (as marked at the github repo). Fortunately we can use pip (a python package manager).
pip install biopython
pip install pybedtools
In the unlikely case you are using a python2 version < 2.7.9 or a python3 version < 3.4, then you may not have pip. But there are helpful instructions for getting it at the pip installation page.
NCBI’s EDirect
If you’re dancing in the bioinformatics world, at some point you will no doubt find yourself wanting to download a massive amount of gene sequences or reference genomes or other information from the glorious NCBI. One of the ways you can download things in bulk from the command line is using their EDirect command-line tools.
It can be tricky to use these, I’ve struggled quite a bit with it, but it’s also very powerful and sometimes the only way to get what I’ve needed. I have some examples up on this page.
Conda install
conda install -y -c conda-forge -c bioconda -c defaults entrez-direct
Non-conda way
This link has the following installation instructions, which I’ve been able to execute successfully on both some Mac and on a Linux systems, but have run into issues on others. Conda has been much more consistently successful for me (that’s pretty much true for every program 🙂).
Copying and pasting these commands into your terminal should do the trick:
cd ~
/bin/bash
perl -MNet::FTP -e \
'$ftp = new Net::FTP("ftp.ncbi.nlm.nih.gov", Passive => 1);
$ftp->login; $ftp->binary;
$ftp->get("/entrez/entrezdirect/edirect.tar.gz");'
gunzip -c edirect.tar.gz | tar xf -
rm edirect.tar.gz
builtin exit
export PATH=${PATH}:$HOME/edirect >& /dev/null || setenv PATH "${PATH}:$HOME/edirect"
./edirect/setup.sh
This downloads and installs EDirect. The last step may take a minute or two, and when it’s done it might tell you a command you need to run in order to add the appropriate directory to your PATH. Copy and paste that command into the terminal, open a new terminal window or run source ~/.bashrc (or source ~/.bash_profile if it had you add it to that file). Then you’re ready to test out that all is well by running esearch -help, and you should hopefully see something like this:
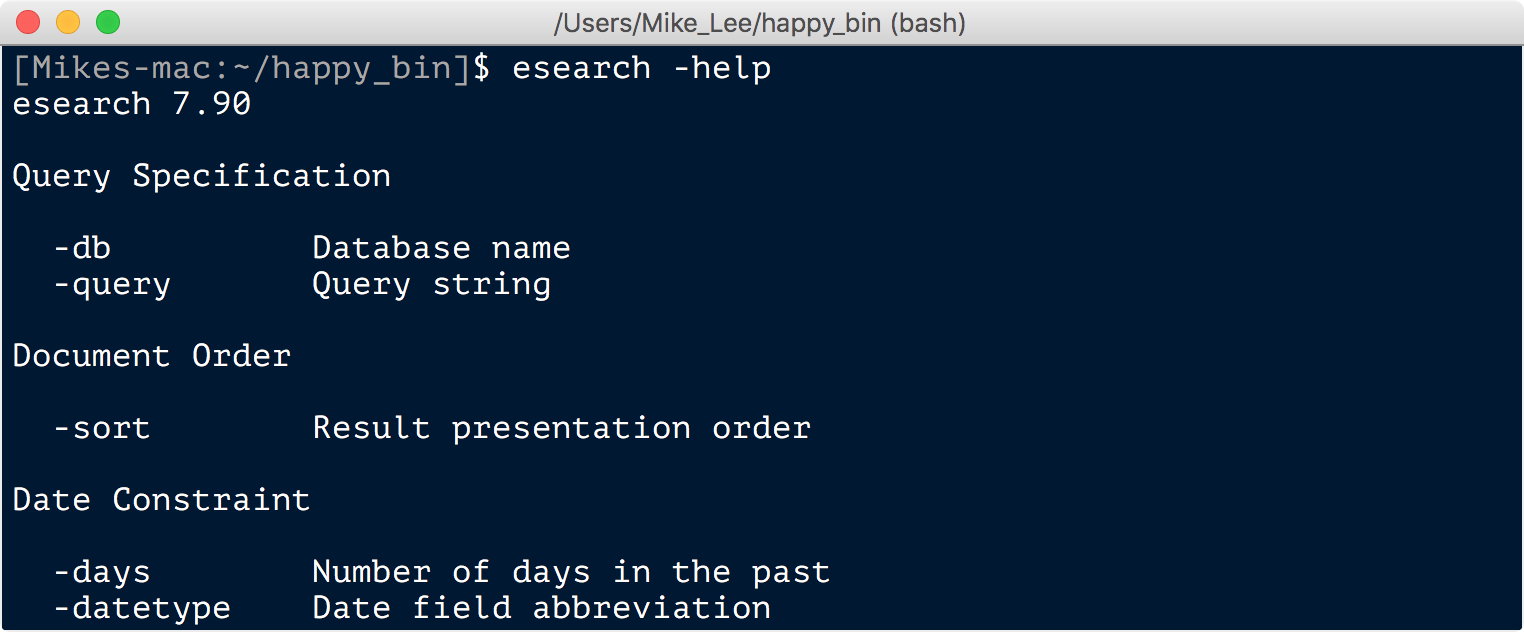
And you can find some example usage here.
vsearch
There are instructions to get vsearch up and running on its github, but these commands should work for you if you’re on a Mac (if you’re not, you’ll have to download a different version you can find following the above link).
cd ~/happy_bin
curl -LO https://github.com/torognes/vsearch/releases/download/v2.5.1/vsearch-2.5.1-macos-x86_64.tar.gz
tar -xzvf vsearch-2.5.1-macos-x86_64.tar.gz
cp vsearch-2.5.1-macos-x86_64/bin/vsearch .
rm vsearch-2.5.1-macos-x86_64.tar.gz
Here we changed into our ~/happy_bin directory, downloaded the vsearch tool with curl, unpacked it and unzipped things with tar, copied the main executable file into our ~/happy_bin directory so that it is in our PATH and can be called from anywhere, then finally we deleted the compressed downloaded file. Lastly, one way we can quickly test that the program seems to be working as it should be is by checking the version:
vsearch --version
Which should return something like this:
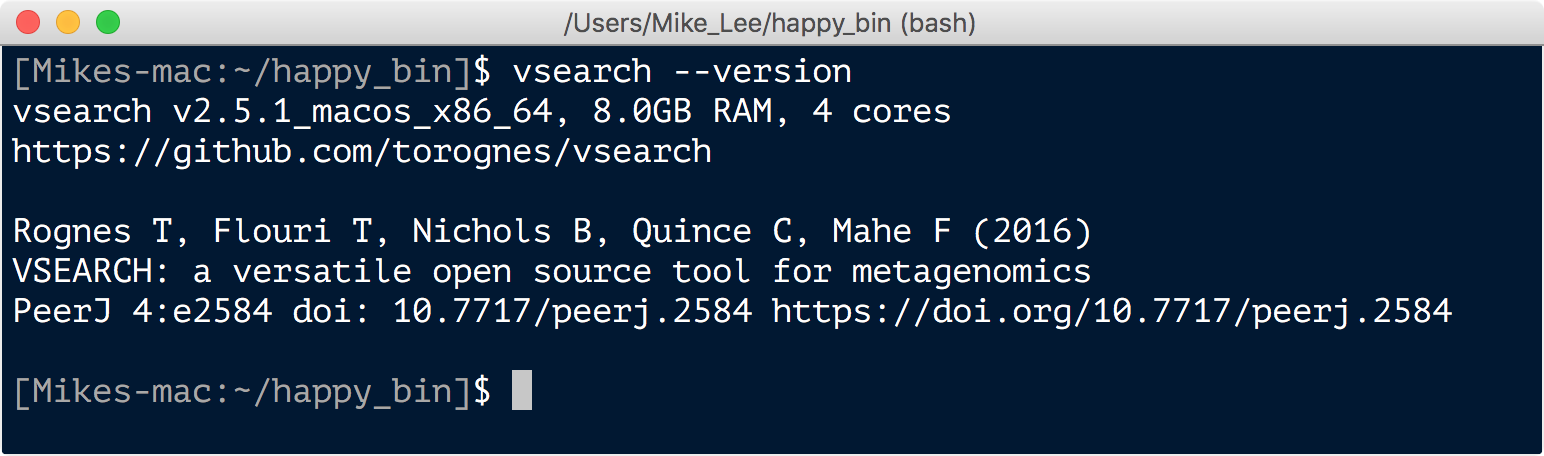
usearch
To get the free version of usearch, you first need to go to https://www.drive5.com/usearch/download.html, and fill out a (very) short form in order to have a download link sent to you. This usually happens virtually instantly. After getting the link and downloading the file, assuming you’re working on a Mac and you downloaded the same version as noted above (v10.0.240) into your default download directory, the following commands will move usearch into our ~/happy_bin directory and change its properties so we can execute it (if you download a different version, adjust the commands accordingly):
cd ~/happy_bin
mv ~/Downloads/usearch10.0.240_i86osx32 usearch
chmod +x usearch
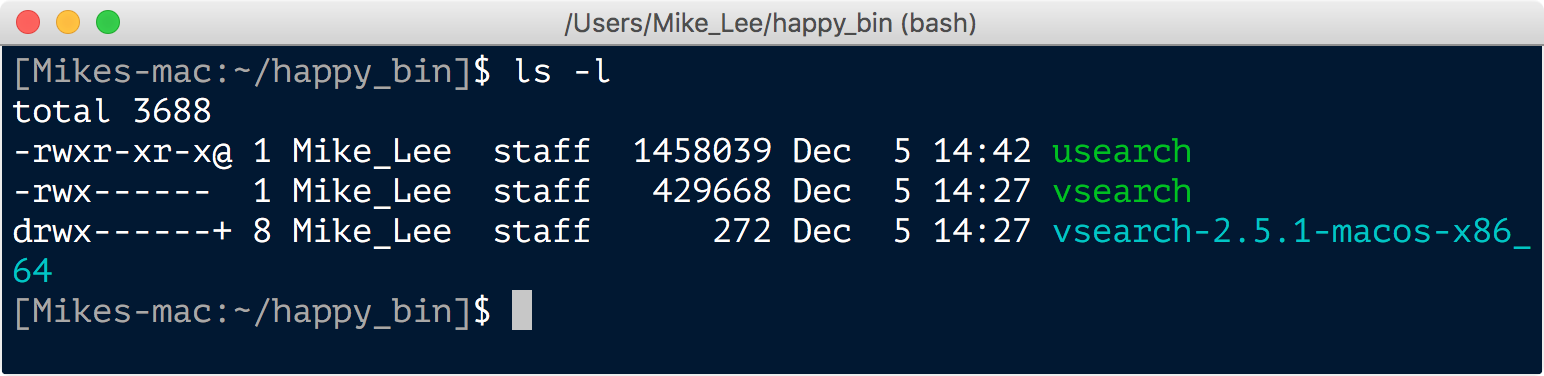
Here we changed into our ~/happy_bin directory, moved the downloaded file from our downloads directory to here and renamed it to simply “usearch” with the mv command, and then ran the chmod command to make the program executable so we can actually run it. Sometimes this chmod step is needed and sometimes it isn’t depending on how the developer packaged the program. If we had tried to run usearch before running the chmod command, we would have gotten an error saying permission denied. We can check the permissions and properties ascribed to a file or directory by using the ls command with the -l optional argument:
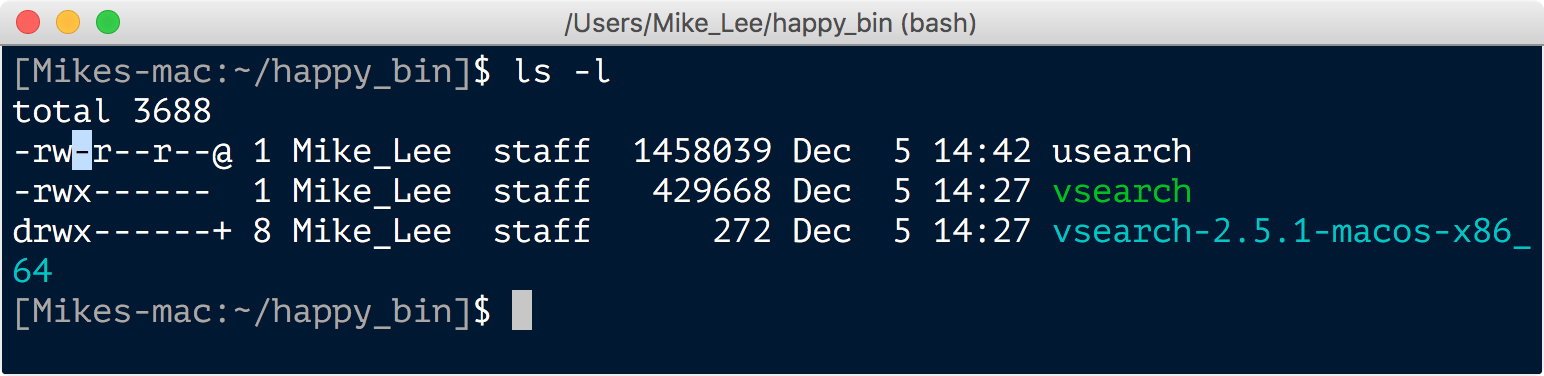
The details of what all of the letters and their positions mean here are a bit further into the weeds than we need to worry about right now, but quickly, an “r” is for readable, a “w” is for writable, and an “x” is for executable. Note that the vsearch executable file has an “x” in the position I highlighted for the usearch file. After running chmod +x usearch, the output of ls -l looks like this:
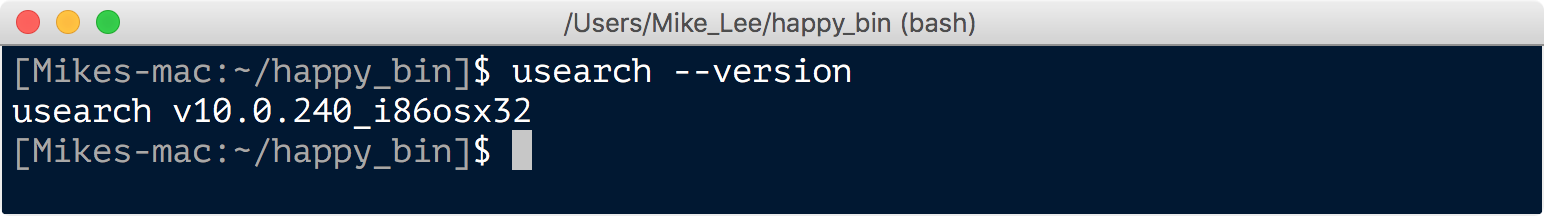
And now the usearch program can be executed (run) without throwing any errors, which we can again check quickly by asking for the version:
usearch --version
FastQC
FastQC is a handy and user-friendly tool that will scan your fastq files to generate a broad overview summarizing some useful information, and possibly identify any commonly occurring problems with your data. But as the developers note, its modules are expecting random sequence data, and any warning or failure notices should be interpreted within the context of your experiment.
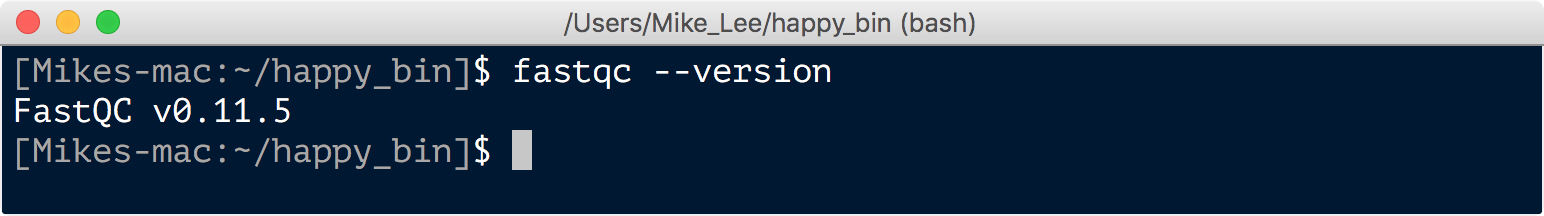
A link to download the current version of fastqc for your system can be found here. And there are pretty good instructions for different platforms provided here, including possibly needing to update your Java installation. For OSX there is a disk image, or application bundle, available to download. So if you’re on a Mac, you can download the “fastqc_v*.dmg” file from the above downloads link, open it to install the program, and then move the resulting “FastQC.app” file (which is actually a directory) into your ~/Applications directory or into your ~/happy_bin if you’d like. I haven’t gone through this on anyone else’s computer, but on mine the installation itself seems adds the executable file to my /usr/local/bin so that we can call it from anywhere. We can check this again by asking for which version:
fastqc --version
And an example usage at the command line to run two fastq files through would look like this:
fastqc sample_A.fastq.gz sample_B.fastq.gz
You can list multiple files like this delimited by a space, and the program is capable of dealing with gzipped files. This will produce a .html file for each fastq file you run it on that you can then open and analyze.
Trimmomatic
Trimmomatic is a pretty flexible tool that lets you trim and/or filter your sequences based on several quality thresholds and some other metrics (e.g. minimum length filtering, or removing adapters). It runs as a java program, so the same binary seems to work across systems. The binary can be downloaded from this link, and here is one way to do this at the command line to grab the current version at the time I’m putting this together:
cd ~/happy_bin
curl -LO http://www.usadellab.org/cms/uploads/supplementary/Trimmomatic/Trimmomatic-0.36.zip
unzip Trimmomatic-0.36.zip
rm Trimmomatic-0.36.zip
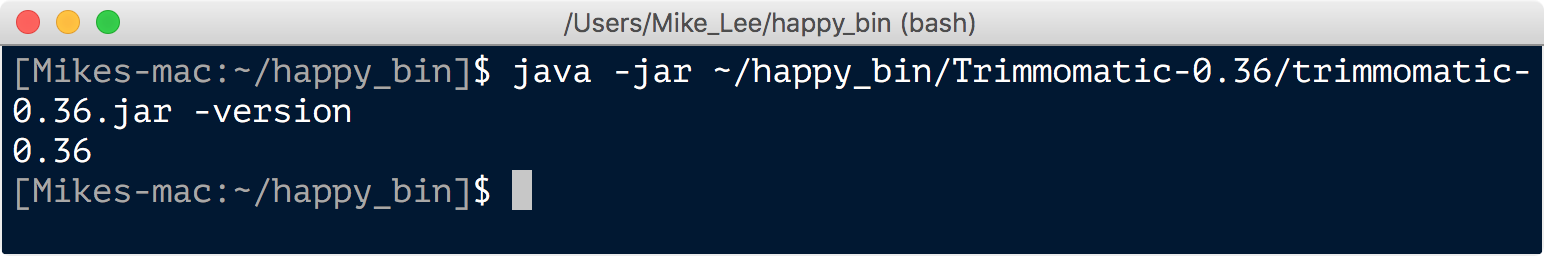
At this point, Trimmomatic, should be ready to rock, and we can again check quickly by asking for the version:
java -jar ~/happy_bin/Trimmomatic-0.36/trimmomatic-0.36.jar -version
I never figured out how to get a .jar to be callable from anywhere without providing the full path like I’ve done here, so this is just how I run it (always providing the full path). If you’re reading this and you do know how to do that, please shoot me a message 🙂
sabre
sabre is an awesomely simple and quick tool for demultiplexing your samples and trimming off the barcodes. The installation seems to run smoothly wherever I’ve tried it, and the usage examples on their github are very straightforward. Here’s how I installed it on my mac:
cd ~/happy_bin
curl -LO https://github.com/najoshi/sabre/archive/master.zip
unzip master.zip
cd sabre-master
make
cp sabre ../
cd ../
sabre # should print help menu
bbtools
Brian Bushnell has made a very handy set of tools called bbtools (also called bbmap). Installation instructions from the developer are available here, but here are steps I used to install it on my MacOS in our ~/happy_bin:
cd ~/happy_bin
curl -L https://sourceforge.net/projects/bbmap/files/latest/download -o bbtools.tar.gz
tar -xzvf bbtools.tar.gz
Since bbtools comes with many programs, stored within the directory we just untarred, it may be preferable to add that directory to our PATH, rather than copying all of the programs to our location here. So here we are adding another directory to our PATH:
cd bbmap/
pwd # copying the full path from the output of pwd to paste it into the following command, your location will be different
echo 'export PATH="$PATH:/Users/Mike_Lee/happy_bin/bbmap"' >> ~/.bash_profile
source ~/.bash_profile # this file is run when we open a terminal session, since we just changed it we need to open a new one or run the `source` command on it for our changes to take effect
cd ~/happy_bin
And then checking the installation using one of the programs:
bbduk.sh --version
If this finds the program and tells you the version (even if there are some memory messages in there), then you’re good to go!
illumina-utils
The illumina-utils library provides a suite of tools for working with Illumina paired-end data put out by merenlab.org. I most commonly use some of the quality filtering programs they make available. A few paths to installation can be found here, which are pretty straightforward other than you do need to be working in a python 3 environment. Since the world is amid the switch from python 2 to python 3, this may complicate things for you in some cases. The easiest way around it I’ve found is working with virtual environments, which I hope to add in soon. For now, since I have a python 3 setup on my computer that I call with python3, and a pip for it that I call with pip3, I can install this way:
pip3 install illumina-utils
You can then see a list of all the programs by typing iu- and hitting tab twice:
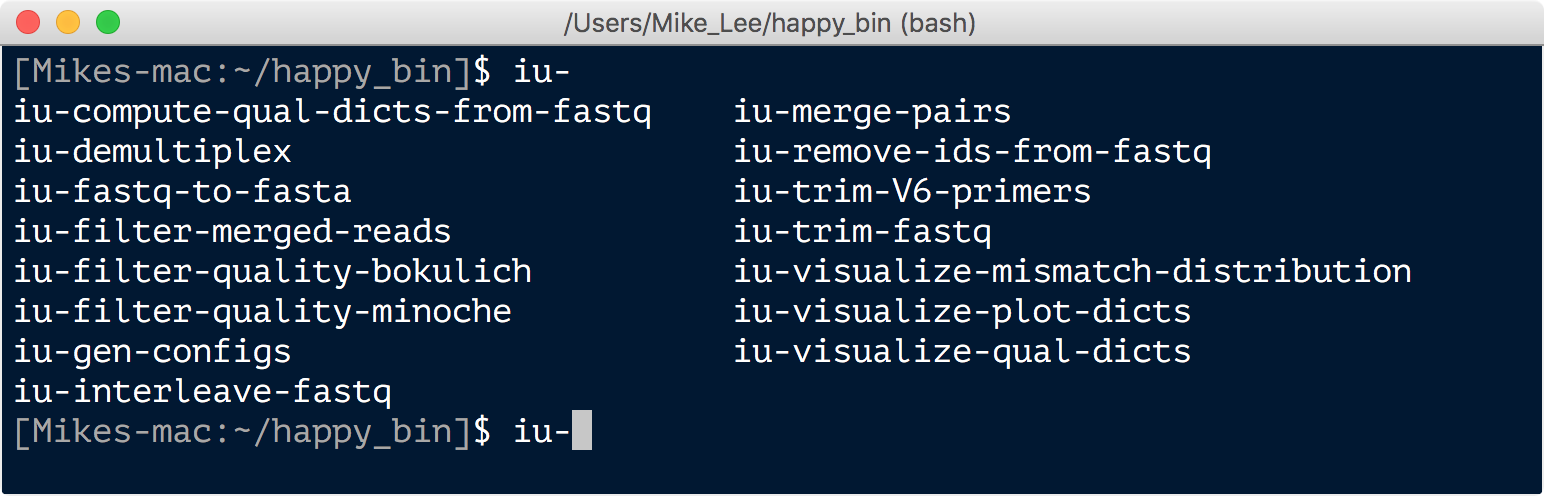
And to be sure things are functioning properly, and the correct version of python is being utilized, we can check for a version:
iu-demultiplex --version
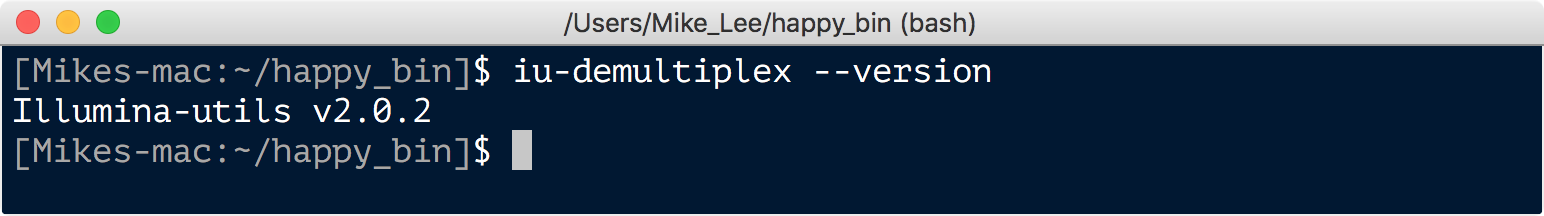
I hope to get to virtual environments soon, I apologize if I’m leaving you hanging on this for the moment!
QUAST
QUAST is a really nice tool for comparing multiple assemblies, and for metagenome assemblies there is a comparable MetaQUAST. Some discussion and example usage can be found here. To install on my personal computer, I followed the instructions laid out here, and, because of the way QUAST compiles things as needed if used, I added its location to my PATH:
cd ~/happy_bin
curl -LO https://downloads.sourceforge.net/project/quast/quast-4.6.1.tar.gz
tar -xzvf quast-4.6.1.tar.gz
rm quast-4.6.1.tar.gz
cd quast-4.6.1
sudo ./setup.py install # will need to enter your password, and actually puts the executable in your /usr/local/bin
cd ~/happy_bin
And testing it’s actually callable:
quast.py --version
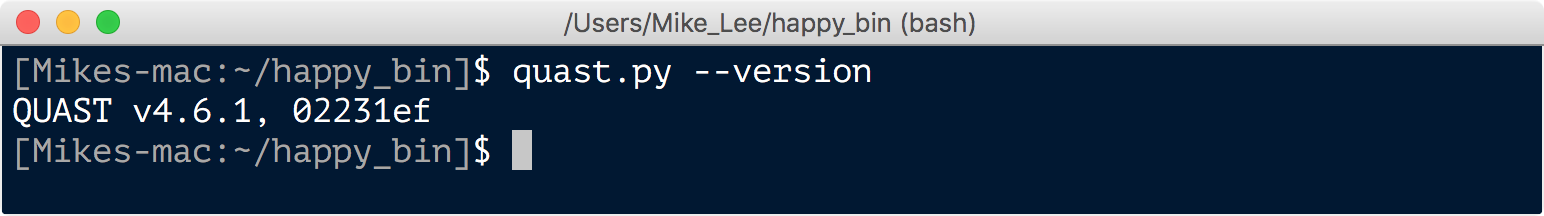
SPAdes
SPAdes is an assembly program. You can read some of my thoughts on assemblies here. SPAdes is packaged as a binary, and the developers provide excellent installation instructions here. This is how I installed the latest release at the time on my local computer:
curl -LO http://cab.spbu.ru/files/release3.11.1/SPAdes-3.11.1-Darwin.tar.gz
tar -xzvf SPAdes-3.11.1-Darwin.tar.gz
rm SPAdes-3.11.1-Darwin.tar.gz
Since SPAdes comes with many programs, stored in a subdirectory of the SPAdes directory we just untarred, it may be preferable to add that directory to our PATH, rather than copying all of the programs to our location here. So here we are adding another directory to our PATH:
cd SPAdes-3.11.1-Darwin/bin/
pwd # copying the full path from the output of pwd to paste it into the following command, your location will be different
echo 'export PATH="$PATH:/Users/Mike_Lee/happy_bin/SPAdes-3.11.1-Darwin/bin"' >> ~/.bash_profile
source ~/.bash_profile # this file is run when we open a terminal session, since we just changed it we need to open a new one or run the source command on it for our changes to take effect
cd ~/happy_bin
And then checking the installation:
spades.py --version
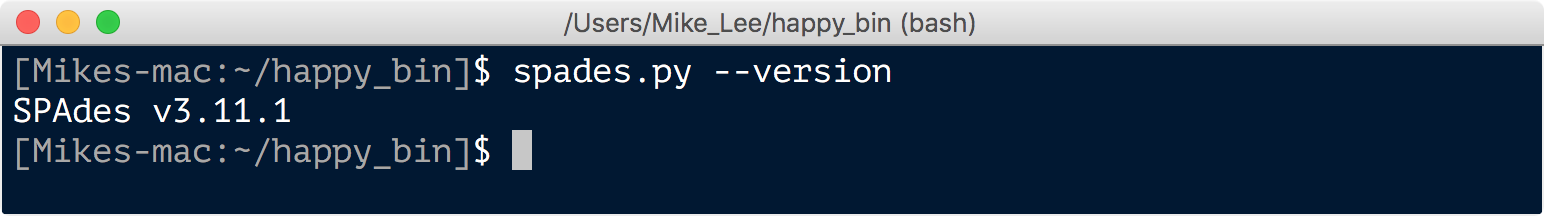
MEGAHIT
MEGAHIT is another assembly program that is great on memory requirements and speed. This can be installed from source, or through git as noted on the above linked page, but there are also binaries available here. As there is an extra layer of complexity due to OSX g++, noted here, I grabbed the latest available binary for Mac OSX, which only seems to be missing one bug fix I haven’t happened to run into:
cd ~/happy_bin
curl -LO https://github.com/voutcn/megahit/releases/download/v1.1.1/megahit_v1.1.1_DARWIN_CPUONLY_x86_64-bin.tar.gz
tar -xzvf megahit_v1.1.1_DARWIN_CPUONLY_x86_64-bin.tar.gz
rm megahit_v1.1.1_DARWIN_CPUONLY_x86_64-bin.tar.gz
cp megahit_v1.1.1_DARWIN_CPUONLY_x86_64-bin/megahit* .
And quick testing, all seems to be well:
megahit --version
Magic-BLAST
NCBI’s Magic-BLAST is a tool based on general BLAST principles but built to deal with high-throughput data, like Illumina reads, considers paired-reads, and can deal with fastq files. It has binaries available for mac, linux, and windows which can be downloaded here. Here’s how I grabbed it:
cd ~/happy_bin
curl -LO ftp://ftp.ncbi.nlm.nih.gov/blast/executables/magicblast/LATEST/ncbi-magicblast-1.3.0-x64-macosx.tar.gz
tar -xzvf ncbi-magicblast-1.3.0-x64-macosx.tar.gz
rm ncbi-magicblast-1.3.0-x64-macosx.tar.gz
cp ncbi-magicblast-1.3.0/bin/* . # moving the executables to our ~/happy_bin so they are in our PATH
magicblast -version # for me, magicblast: 1.3.0